文獻解讀:干預內皮細胞PFKFB3或CtBP1活性可以緩解VR提供新干預靶點
血管重構(VR)是以血管阻力增加、內膜增厚、節段性狹窄或動脈瘤樣擴張為特征的復雜的結構性病理改變,是多種心血管疾病(CVDs)的重要病理性標志。血管內皮細胞(ECs)通過感知和轉導誘導重構的刺激,產生調節細胞生長、凋亡和細胞外基質組成的介質,促進血管重塑。內皮間充質轉化(EndoMT)作為一種內皮異常狀態,廣泛參與動脈粥樣硬化和心臟纖維化等疾病,但其在VR的作用和機制尚未闡明。糖酵解(Glycolysis)過程在內皮多種病理狀態下功能實現中發揮了關鍵作用,但其對EndoMT及VR發生發展的作用和調控機制尚不清楚。
2023年10月5日,廣州醫科大學基礎醫學院心血管平臺徐益鳴教授團隊在Hypertension(醫學1區,IF=8.3)上發表了題為“Glycolysis Promotes Angiotensin II-Induced Aortic Remodeling Through Regulating Endothelial-to-Mesenchymal Transition via the Corepressor C-Terminal Binding Protein 1”的研究論文,揭示了內皮細胞中PFKFB3介導的糖酵解對EndoMT激活的全新調控通路及其在VR進程中的關鍵作用,為抗VR治療提供了全新的代謝干預靶點。該論文的通訊作者為徐益鳴教授,第一作者為復旦大學附屬中山醫院博士生王立濤和廣州醫科大學基礎醫學院博士生郭帥。

研究材料
在此項目中,研究人員構建了內皮特異性PFKFB3敲除小鼠模型(Pfkfb3iECKO)探究內皮PFKFB3對EndoMT激活及VR進程的調控作用。隨后使用AAV病毒(由賽業生物提供)實現了在內皮中對CtBP1的點突變,并進行了詳細的下游機制驗證。
研究方法
在此項目中研究人員采用了多項在體和體內實驗技術,包括en face免疫熒光、主動脈HE、SR和EVG染色、Western blotting及RT-PCR等。在探究CtBP1與E2F4的結合對SMURF2的轉錄調控作用時,研究人員使用了CHIP及熒光素酶報告基因實驗。此外,研究人員還測量了胞內NADH和NAD+的水平。
技術路線
01 VR中EndoMT與PFKFB3的表達相關性
02 體內外抑制PFKFB3對EndoMT及VR進程的影響
03 PFKFB3調控EndoMT過程的分子機制
04 CtBP1/E2F4轉錄復合物對EndoMT和VR的調控作用及其機制
研究結果
1 VR病理狀態下EndoMT過程和內皮TGF-β1/SMAD2信號被激活
首先研究人員通過Ang II皮下緩釋誘導的小鼠血管重塑模型。為了評估EndoMT在血管重塑中的發生情況,選取省里鹽水或Ang II緩釋7天的小鼠胸主動脈進行檢測。En face免疫熒光染色顯示,與生理鹽水處理的小鼠相比,Ang II緩釋后VE-cadherin(ECs標志物)的表達明顯減少,而Vimentin(間充質細胞標志物)的表達明顯增加,提示EndoMT過程被激活。此外,在Ang II緩釋的小鼠胸主動脈內皮層觀察到TGF-β1信號中間體SMAD2的磷酸化增加。以上結果表明,在Ang II誘導的血管重塑過程中,內皮TGF-β/SMAD2信號和EndoMT過程被激活。
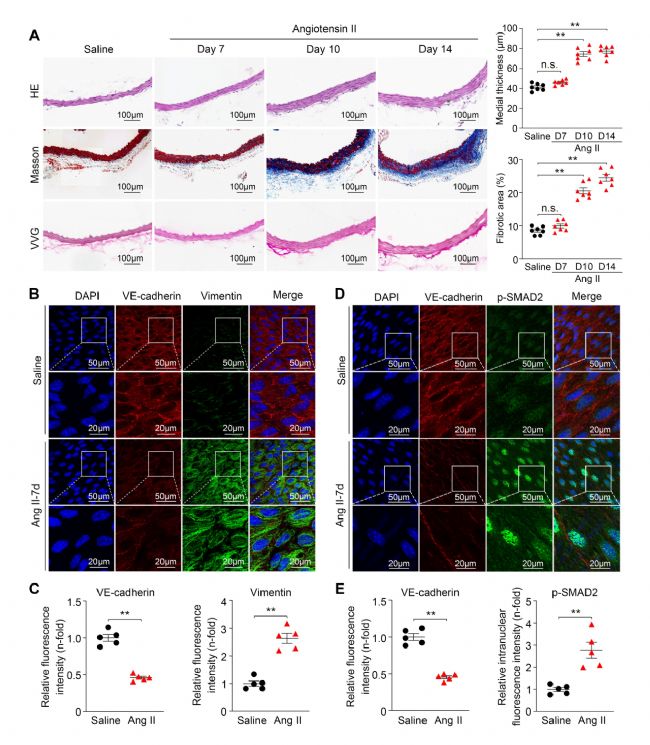
圖1 VR病理狀態下EndoMT過程和內皮TGF-β1/SMAD2信號被激活[1]
2 EndoMT過程伴隨PFKFB3介導的糖酵解增加
隨后,研究人員發現Ang II緩釋的小鼠主動脈內皮中糖酵解關鍵酶PFKFB3的表達顯著升高。通過使用TGF-β1刺激人臍靜脈內皮細胞(HUVECs)誘導EndoMT,細胞明場拍照結果顯示TGF-β1處理使HUVECs表現為成纖維或間充質細胞的形態,同時細胞的伸長指數也顯著升高。此外,與動物水平的結果一致,TGF-β1體外處理顯著增加了糖酵解關鍵酶PFKFB3的表達水平。海馬細胞外通量分析也提示TGF-β1處理的HUVECs中基礎糖酵解、糖酵解能力、最大糖酵解和糖酵解儲備均有所增加;基因敲除PFKFB3能顯著降低TGF-β1誘導的細胞糖酵解水平升高。
為了進一步探究TGF-β1誘導PFKFB3蛋白表達升高的機制,研究人員使用JASPAR軟件分析了PFKFB3的啟動子區域,并發現了兩個SMAD2/3結合元件(SMAD2/3 binding elements, SBEs)。進一步使用抗SMAD2/3的抗體進行染色質免疫共沉淀(ChIP)實驗發現,SMAD2/3能夠與PFKFB3上包含SEBs的啟動子區域結合,而且這種結合在TGF-β1處理后顯著增加。此外,熒光素酶報告基因實驗表明,SMAD2/3的過表達以劑量依賴性的方式顯著增加PFKFB3的啟動子活性,進一步證明PFKFB3是SMAD2/3復合物的轉錄靶標。
綜上所述,在EndoMT過程中,TGF-β1/SMAD2信號通過轉錄激活PFKFB3基因,誘導內皮糖酵解升高。

圖2 EndoMT過程伴隨PFKFB3介導的糖酵解增加[1]
3 內皮PFKFB3介導EndoMT和VR形成
為了剖析內皮PFKFB3上調在Ang II誘導的血管重塑中的功能性作用,研究人員構建了他莫昔芬誘導的內皮特異性Pfkfb3敲除(Pfkfb3iECKO)和對照組(Pfkfb3fl/fl)小鼠。將Pfkfb3fl/fl和Pfkfb3iECKO小鼠經Ang II皮下緩釋14天后,進行組織學染色和分析。而經Ang II緩釋后,Pfkfb3fl/fl小鼠胸主動脈的中膜和外膜膠原含量、中膜厚度、以及外層彈性纖維(EEL)周長顯著增加;然而,所有這些病理特征在Pfkfb3iECKO小鼠中都得到改善。此外,en face染色顯示PFKFB3缺失顯著抑制了Ang II誘導的EndoMT過程和p-SMAD2水平的升高。以上結果表明內皮PFKFB3缺乏能夠減輕Ang II誘導的主動脈重塑和EndoMT。

圖3 內皮PFKFB3介導EndoMT和VR形成[1]
4 NADH依賴性的CtBP1激活對于EndoMT的誘導至關重要
此前的證據表明NADH/NAD+比率增加與糖酵解的熱力學耦合,在基因轉錄中發揮著根本性的作用。研究人員首先發現,TGF-β1處理顯著提高HUVECs中的NADH含量和NADH/NAD+比率,基因敲除或PFK158藥物抑制PFKFB3則能逆轉這一變化,但均不影響NAD+的水平。為了明確NADH/NAD+比率增加與EndoMT之間的功能關系,研究人員使用4-甲硫基-2-氧代丁酸(MTOB)耗竭胞內NADH,結果發現逆轉了ECs中間充質細胞標志物的上調和內皮標志物的下調。羧基末端結合蛋白1(CtBP1)是一種眾所周知的NADH敏感型的蛋白,并被證實可作為轉錄共抑制因子調控基因轉錄。SiRNA敲除CtBP1能夠模擬PFKFB3缺失的抑制EndoMT效應。隨后,研究人員通過構建對NADH不敏感的CtBP1突變體(CtBP1G183A),發現過表達CtBP1G183A幾乎完全逆轉TGF-β1誘導的EndoMT過程(其中使用的AAV9-CDH5>Ctbp1(WT)和AAV9-CDH5>Ctbp1(G183A)病毒由賽業生物提供)。綜上所述,這些數據表明PFKFB3驅動的糖酵解以NADH/CtBP1依賴性的方式正向調控EndoMT。
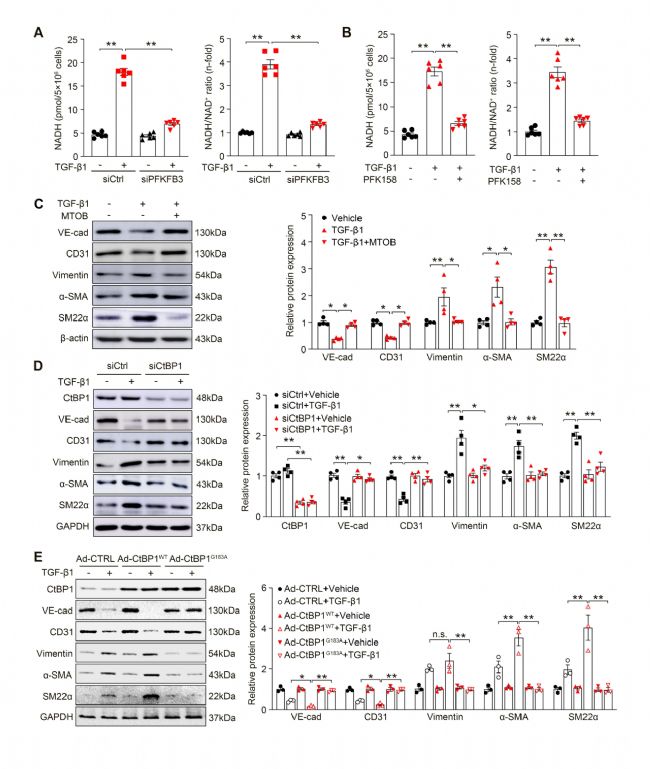
圖4 NADH依賴性的CtBP1激活對于EndoMT的誘導至關重要[1]
5 PFKFB3/NADH/CtBP1軸通過抑制SMURF2促進EndoMT
為了更深入地探究糖酵解依賴性的CtBP1激活如何調控EndoMT過程,研究人員發現敲除PFKFB3或CtBP1均能夠減輕TGF-β1誘導的SMAD2磷酸化。隨后,通過篩選一系列經典的TGF-β1/SMAD2信號的調控因子,研究人員篩選出SMAD泛素調節因子2(SMURF2)是潛在的PFKFB3/NADH/CtBP1軸的作用靶點。過表達野生型CtBP1WT能夠劑量依賴性降低SMURF2的表達;然而,過表達突變型CtBP1G183A則逆轉了對SMURF2的抑制作用。敲除SMURF2能夠消除PFKFB3缺失對SMAD2信號的去激活作用。綜上所述,PFKFB3/NADH/CtBP1軸通過抑制SMURF2的表達激活TGF-β1/SMAD2信號通路。
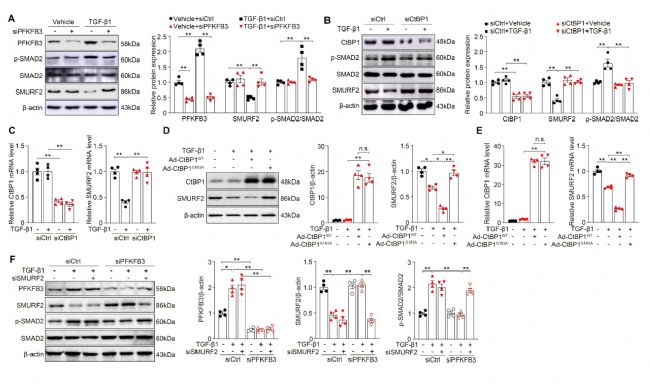
圖5 PFKFB3/NADH/CtBP1軸通過抑制SMURF2促進EndoMT[1]
6 CtBP1/E2F4復合物轉錄抑制SMURF2的表達
CtBP1作為轉錄共抑制因子,既往研究報道CtBP1介導的轉錄抑制功能依賴于CtBP1與其他轉錄抑制因子的結合。研究人員通過大數據篩選發現E2F4是CtBP1發揮抑制作用潛在的轉錄抑制因子。免疫共沉淀(Co-IP)和Re-ChIP實驗共同表明CtBP1與E2F4能夠直接結合,并形成轉錄復合物與SMURF2啟動子結合。PFKFB3敲除或MTOB處理能夠抑制以上結合。熒光素酶報告基因實驗則進一步提示E2F4過表達抑制能夠SMURF2啟動子區域的活性,野生型(WT)而非突變型(G183A)CtBP1的過表達明顯加強TGF-β1處理下E2F4的抑制作用。這些數據表明CtBP1與E2F4結合共同轉錄抑制了SMURF2在TGF-β1處理下的表達。
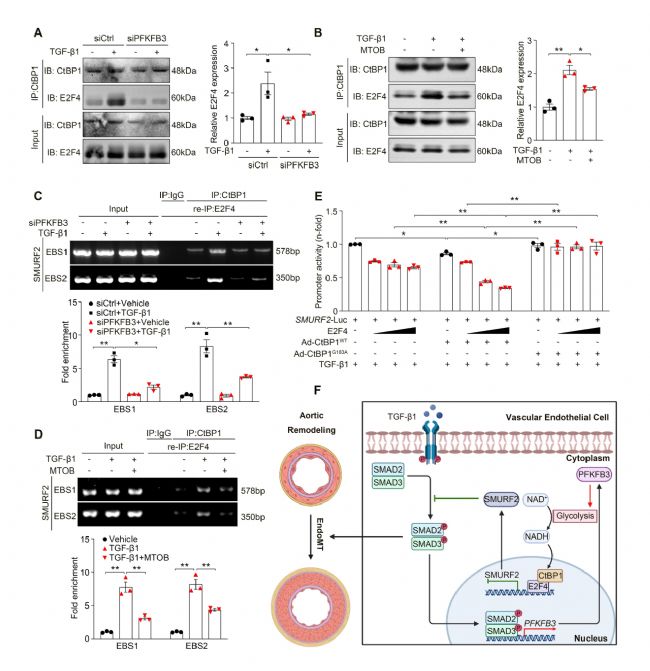
圖6 CtBP1/E2F4復合物轉錄抑制SMURF2的表達[1]
研究結論
此項研究首次證實內皮PFKFB3介導的糖酵解通過調控EndoMT促進VR過程。機制探索表明糖酵解依賴性的NADH水平升高介導了CtBP1/E2F4轉錄復合物對TGF-β1/SMAD2信號負性調控因子SMURF2的轉錄抑制,進而促進EndoMT和VR。此項研究也首次闡明干預內皮細胞PFKFB3或CtBP1活性可以緩解VR,為VR的防治提供了新的干預靶點。

圖7 機制圖[1]
參考文獻:
[1]Wang L, et al. Glycolysis Promotes Angiotensin II-Induced Aortic Remodeling Through Regulating Endothelial-to-Mesenchymal Transition via the Corepressor C-Terminal Binding Protein 1. Hypertension. 2023 Dec;80(12):2627-2640. doi: 10.1161/HYPERTENSIONAHA.123.21382.