
羅氏NimbleGen序列捕獲技術實現轉錄組定向測序
最近在Nature Protocols雜志上在線發表的一篇文章1介紹了利用NimbleGen序列捕獲技術進行轉錄組定向測序,用于RNA研究中基因發現和表達定量。
RNAseq技術在基因表達研究中得到了廣泛的應用,可對表達基因進行無偏見的采樣。相比定量PCR可以對更廣泛的基因同步分析,相比芯片方法RNAseq測序結果準確性也更高。然而,真核細胞的表達譜具有轉錄本數量眾多、可變剪切情況復雜,且表達量高低差異大的特點。RNAseq測序結果分散在整個基因組中,不同基因測序深度差異顯著,對轉錄水平偏低的轉錄本往往測序深度不足,為進行特定基因的可變剪切體拼接及定量研究造成阻礙。
而序列捕獲技術通過寡合苷酸探針雜交,富集研究所感興趣的基因或基因組區段,實現了定向測序。將此技術與RNAseq結合,即對RNAseq的測序文庫先進行序列捕獲實驗富集感興趣的轉錄本,再進行測序,實現定向RNAseq或RNA捕獲測序(CaptureSeq)。這一方法可以提高目標轉錄本的測序深度,進行靈敏的基因發現、有效的轉錄本組裝和準確的基因表達定量。
文中介紹的實驗步驟主要包括探針設計、捕獲測序和數據分析如轉錄本拼接及定量。實驗選用了羅氏NimbleGen探針用于目標捕獲。NimbleGen可同時生產2萬種探針,可覆蓋多達200Mb的基因組中的分散區域或連續的區域,可以應對復雜的真核生物轉錄組。文章指出,在探針設計時,僅針對目標轉錄本的部分序列設計探針,通過這些探針即可捕獲全長轉錄本,也可發現轉錄本中的新外顯子。當需對特定剪切方式的轉錄本進行定向研究時,可在探針設計包括連接兩個外顯子片段的探針,專門捕獲這些異構體。
此外,實驗設計了用于質控的探針。質控探針包括:1. 非轉錄的基因間區的探針,以排除gDNA污染;2. 其他可能造成實驗室污染如大腸桿菌等的序列捕獲探針,以排除實驗室污染;3. 數個質控基因的探針,可用于進行測序前qPCR富集分析,或用于數據分析參數的測試;4. 針對摻入樣本的ERCC RNA設計的探針。 ERCC RNA Spike-in standards由92個in vitro轉錄的轉錄本組成,表達量跨越一個106的范圍,這一些標準品被摻入到最初的RNA樣品中進行捕獲測序,用于判斷測序量是否充足,以及計算捕獲off-target rate。另外,將各轉錄本獲得的測序深度,與ERCC RNA Spike-in standards的測序深度比較,可以推算出樣本中各轉錄本的數量。文章作者指出,這個方法應用于轉錄水平較低的基因的富集,富集率與基因數量相關,如1000個隨機選擇的基因預期55倍富集效率。
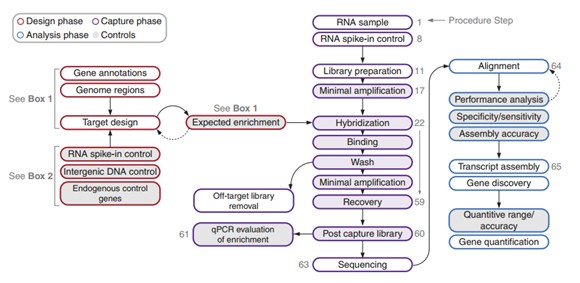
圖:定向RNAseq測序(CaptureSeq)實驗流程。摘自:Tim R Mercer et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nature Protocols.2014 doi:10.1038/nprot.2014.058.
5微克的總RNA約可生成250ng cDNA文庫,可將多個樣本的文庫混合后根據Roche NimbleGen的實驗手冊進行捕獲實驗。混合捕獲的實驗方案可以對大量樣本進行低成本的測序。測序后的分析步驟包括比對、拼接、去除非目標序列、新基因(外顯子)發現、定量等。
通過這一實驗,可以對樣本中特定基因的表達進行研究。該方法既RNASeq相比其他表達譜研究方法的優勢,又可以進行有目的性的、多樣本的研究。尤其在對于表達水平中低等的轉錄本研究中,可以更好地拼接全長轉錄本、發現新基因以及定量分析。
1. Tim R Mercer, Michael B Clark, Joanna Crawford, Marion E Brunck, Daniel J Gerhardt, Ryan J Taft, Lars K Nielsen, Marcel E Dinger, John S Mattick. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nature Protocols.9, 989–1009 (2014) doi:10.1038/nprot.2014.058.
僅用于科研,不用于診斷。


- 百蓁生物邀您共赴CBI 2025生物醫藥創新博覽會
- 明美光電首屆蘇州經銷伙伴大會圓滿舉辦
- 中國醫科大學功能超聲fUS成像儀ICONEUS ONE成功裝機
- 上海優耳儀器科技有限公司成立20周年答謝函!
- 覃思科技成為丹麥DeltaPix顯微成像產品中國區代理商
- 易科泰創新實踐國家發改委“百城千企”課題
- 盧湘儀離心機亮相央視,國產科研利器賦能經濟發展
- 贊德公司關于美國Med associaties中國獨家代理聲明
- MCE推出AI藥物篩選平臺,實現數千萬的分子快速篩選
- 迪必爾入選2025關鍵技術研發計劃"合成生物學"項目
- 美國國立衛生研究院宣布停止對僅動物研究的資助
- 智聽自然,聲動未來:沃德精準亮相"聲景中國"研討會
- 赫西智能高速冷凍離心機亮相央視"革新人工催產技術"
- 易科泰榮膺“SFAA 植物工廠應用十大優秀企業”稱號
- 百蓁生物推出一站式高精度HDX-MS分析服務
- 百蓁生物邀您共赴CBI 2025生物醫藥創新博覽會
- 第八屆腫瘤免疫及伴隨診斷合作峰會通知
- PacBio推出采用HiFi技術的桌面型長讀長測序儀Vega
- 因美納邀您參加CSCO第九屆血液腫瘤學術大會
- NGDx2025中國先進診斷技術開發與應用論壇議程表
- 10x Chromium GEM-X動物及畜牧類研究獎勵計劃啟動
- Vizgen攜森西賽智亮相空間生物國際會議分享空轉進展
- 怡美通德正式成為PacBio中國區授權代理商
- 相約BioCon2025,因美納邀您共話腫瘤精準醫學新篇章
- 怡美通德邀您共赴單細胞及空間轉錄蛋白多組學論壇
- 10x Genomics技術方案更新(5月),新手冊邀您下載
- 10x新品發布網絡研討會:Visium HD 3’空間分析方案
- Oxford Nanopore明日直播:納米孔測序大會亞太專場
- 10x邀您報名Visium和Xenium數據分析研討會(北京場)
- 第三屆中國國際生殖醫學健康大會(北京站)通知



Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com